
Module 1 lab index | Next lab day >
Introduction
Back when he was a postdoctoral fellow, Professor Niles screened a random library of RNA aptamers to find one that binds to heme – the iron-containing site in hemoglobin. It is known that heme can bind to certain transcription factors and modulate gene expression (see references Zhang and Hach, 1999 and Ogawa, et al., 2001), and RNA aptamers are one potential tool for learning more about signaling networks involving heme. To select heme-binding aptamers, Professor Niles ran a pool of RNAs with 50 randomized base pairs through a heme affinity column. He then amplified the column-selected pool of aptamers and repeated the process several times. An aptamer called "6-5" survived through the 6th round of screening, but ultimately was found not to bind to heme. An aptamer called "8-12" survived through all 8 rounds of screening, and has a heme binding affinity of 220 nM. Both were described in the Niles, et al., 2006 paper referenced below.
Today you will be given two archival plasmids containing the 6-5 and 8-12 sequences, respectively. RNA is not very stable compared to DNA; thus, RNA aptamers are copied into their associated DNA sequences for long-term storage. Ligating the DNA fragment into a plasmid that can be carried in bacteria provides further amplification and storage capabilities. We will make use of these capabilities more extensively in Module 2.
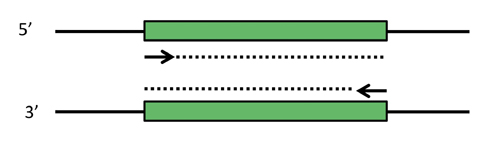
PCR schematic. Depicted are two complementary strands of DNA, with a desired target fragment shown in green. Primers that can select the target sequence are shown as short arrows, with the dotted lines indicating the extension step of PCR. Note that in the first couple rounds of PCR, products longer than the desired target will be made (dotted lines keep extending). However, these early products themselves become templates that produce the correct product in abundance.
In order to select and amplify just the short DNA fragment that encodes for the aptamer, you will use the polymerase chain reaction, PCR. PCR comprises three main steps: 1) template DNA containing a desired sequence is melted, 2) primers anneal to specific locations on the now melted (i.e., single-stranded) DNA, and 3) the primers are extended by a polymerase to select and create the desired product. Extension occurs at ~70 °C, melting at ~95°C, and annealing at a temperature ~5 °C below the primer melting temperature; thus, the repetition of these steps is called thermal cycling. After each cycle, the newly formed products themselves become templates, causing exponential amplification of the selected sequence. (Note that the early rounds of PCR will not produce the desired product - we will see why in today's pre-lab lecture.)
Once the PCR is running, you will begin to explore some computational tools for RNA analysis. During this module, you will ultimately use three different programs to explore both sequence similarities among RNA candidate aptamers and higher-order structures that arise from the primary sequences. For today, you will look at degrees of sequence similarity among a list of aptamers, some of which bind to heme and some that don't.

(Photo by Mark Robert Halper. Courtesy of Kary Mullis. Used with permission.)
Based on the numerous applications of PCR, it may seem that the technique has been around forever. In fact it is only 25 years old. In 1984, Kary Mullis described this technique for amplifying DNA of known or unknown sequence, realizing immediately the significance of his insight.
"Dear Thor!," I exclaimed. I had solved the most annoying problems in DNA chemistry in a single lightening bolt. Abundance and distinction. With two oligonucleotides, DNA polymerase, and the four nucleosidetriphosphates I could make as much of a DNA sequence as I wanted and I could make it on a fragment of a specific size that I could distinguish easily. Somehow, I thought, it had to be an illusion. Otherwise it would change DNA chemistry forever. Otherwise it would make me famous. It was too easy. Someone else would have done it and I would surely have heard of it. We would be doing it all the time. What was I failing to see? "Jennifer, wake up. I've thought of something incredible." --Kary Mullis from his Nobel Lecture, December 8, 1983.
(Copyright © The Nobel Foundation 1993. All rights reserved. This content is excluded from our Creative Commons license. For more information, see http://ocw.mit.edu/fairuse.)
References
Zhang, L., and A. Hach. "Molecular Mechanism of Heme Signaling in Yeast: the Transcriptional Activator Hap1 Serves as the Key Mediator." Cell Mol Life Sci 56, no. 5-6 (October 30, 1999): 415-26.
Ogawa, K., et al. "Heme Mediates Derepression of Maf Recognition Element Through Direct Binding to Transcription Repressor Bach1." EMBO J 20, no. 11 (January 1, 2001): 2835-43.
Niles, J. C. "Utilizing RNA Aptamers to Probe a Physiologically Important Heme-Regulated Cellular Network." ACS Chem Biol 1, no. 8 (September 19, 2006): 515-24.
Protocols
Part 1: Lab Practical
You and your partner may work together on the lab practical. (Note: this will not be the case for future quizzes.) You are of course welcome to give different answers should you disagree.
Part 2: Amplify DNA Fragment Encoding Aptamer
Before starting today's wet lab work, you may want to wipe down your pipettes and your benchtop with 70% ethanol.
- You will be given plasmids encoding the DNA for two aptamers: "6-5" and "8-12," both at ~250 μg/mL. Make a plan for adding 2.5 ng of DNA to each PCR reaction, in no more than 20 μL final volume of plasmid and water.
- Note that you may need to serially dilute the DNA. For example, if you need to dilute the DNA 10,000 times to reach a concentration of 2.5 ng/20 μL, then you should make a 1:100 dilution of the original stock, then a 1:100 dilution of the resulting solution. It's best to pipet at least 5 μL of solution at a time, to avoid the inaccuracies associated with pipetting very small volumes. Feel free to run your plan by the teaching faculty.
- Per reaction, add 20 μL of PCR Master Mix, 5 μL of each primer, the DNA template, and water such that the total volume of the reaction is 50 μL. In addition to the two DNA-containing reactions, you should prepare a no-template control that contains pure water without any plasmid.
- What would it mean if your no-template control resulted in amplification of DNA the same size as your aptamers?
- The PCR will run for about 1 hour, on the following cycle:
| SEGMENTS | CYCLES | TEMPERATURE (° C) | TIME |
|---|---|---|---|
| 1 | 1 | 94 | 4 min |
| 2-4 | 25 | 94 | 30 sec |
| 57 | 30 sec | ||
| 72 | 30 sec | ||
| 5 | 1 | 72 | 10 min |
| 6 | 1 | 4 | indefinite |
Part 3: Introduction to Computational Analysis
Your understanding of this module will in part be evaluated by the RNA Computational Analysis assignment. Although you will not be prepared to understand the entire assignment today - until you have a better grasp of what SELEX entails - you should be able to get a good start on the first section.
- Begin by reading the assignment overview and background information. (It's fine if the latter doesn't make perfect sense yet.)
- Download the associated data file and read the background. Again, the introductory text may make more sense in a week or two.
Primer Analysis
Note that all the sequences in the data file are shown 5' to 3'.
- Begin by copying the sense strand of the SELEX library-encoding DNA to a new Word document.
- Be sure to stick with Courier font so each base is the same size.
- Determine and write out the complementary antisense strand (from 3' to 5') directly below the sense strand.
- Which strand will the 5' primer anneal to? Copy and paste to align the primer with said strand – show it either above or below the two strands of template DNA, not in between them. Highlight the primer in bold.
- Which strand will the 3' primer anneal to? Align this primer with said strand and highlight in bold. Be sure that the complementary base pairs line up. What did you have to do to the primer to make this happen?
- Turn in your primer diagram (and answers to the questions above) with your notebook today.
Sequence Alignment
- Today you will work on part one of the computational assignment, namely sequence alignment using the CLUSTAL program. You may not not be able to address all of the interpretive questions yet, but you can begin answering the more straightforward analytical ones such as parts of 2, 3, and 4.
- To capture the screen shot for the required figure, you can use the Grab utility on a Mac.
- Search for "Grab" using spotlight (the magnifying-glass-looking icon in the upper right corner) or go to Applications → Utilities → Grab.
- You are not required to record your work on this part in your notebook.
For Next Time
- Continue to familiarize yourself with the workings of OpenWetWare. Specifically, you should:
- Add your user page to the class's People page..
- Complete the student registration/questionnaire to turn in next time..
- The major assessment for this module will be the RNA Engineering Report. Start to familiarize yourself with its expected structure and content.
- One week from today in the Module 1 Day 3 session, we will have an in-class discussion about an article from the primary scientific literature. You might begin reading and thinking about the paper now, rather than trying to do so only in the two days between Day 2 and Day 3.
Reagent List
- PCR Master Mix (2.5X)
- 62.5 U/ml Taq DNA Polymerase
- 125 mM KCl
- 75 mM Tris-HCl, pH 8.3
- 3.75 mM Mg(OAc)2
- 500 μM each dNTP
- Forward primer: 20μM stock
- Reverser primer: 20μM stock
- Templates: 8-12 and 6-5