
Module 2 lab index | Next lab day >
Introduction
Contrary to how it may be taught in some laboratory classrooms, the process of scientific inquiry encompasses much more than the collection and interpretation of data. A key part of the process is design — of experiments that specifically address a hypothesis and of new materials or technologies. Moreover, any design is subject to continued revision. You might redesign an experiment or tool based on your own own research, or you might consult the vast body of scientific literature for other perspectives. As the old graduate student saying goes, "A month in the lab might save you a day in the library!" In other words, although the process of combing the literature can be arduous or even tedious at times, it beats wasting a month of your time repeating experiments already proven not to work or reinventing the wheel.
During this module, each of you will design and test a new version of inverse pericam (IPC). Today, we will refer to a few primary research articles in order to familiarize ourselves with this recombinant protein and its constituent parts. The fluorescent component of IPC is an enhanced yellow fluorescent protein (abbreviated EYFP), one of the many derivatives of green fluorescent protein (GFP). GFP is naturally produced by jellyfish and was cloned into other organisms in the early 1990's. It has since been exploited as a genetically encodable reporter and mutagenized to vary its excitation and emission spectra. The other key component of inverse pericam is the protein calmodulin (CaM), a natural calcium sensor that is present in all eukaryotes (and briefly reviewed in Chin and Means (2000)). Calmodulin has many ligands that it binds only in the presence of calcium ion, including the peptide fragment M13. This conditional specificity for M13 binding is enabled by the change in CaM's conformation when it binds calcium.
Within inverse pericam, M13 and CaM are located at opposite ends, surrounding a permuted (i.e., rearranged) version of EYFP. In the absence of calcium, this EYFP exhibits strong fluorescence. However, when calcium is added to a solution of inverse pericam, CaM and M13 interact, disrupting the conformation and, as a result, the fluorescence of EYFP. The transition from bright to dim fluorescence occurs over a particular concentration range of calcium. Your goal today is to propose a mutation that will shift the concentration range over which IPC fluorescence decreases. Specifically, you will modify the calcium sensor portion of inverse pericam in a manner that is likely to increase or decrease its affinity for calcium ion.
In order to make reasonable modifications to inverse pericam, we will use several protein analysis tools. Proteins are modular materials that may be described and examined at multiple levels of a structural hierarchy (from primary to quaternary in the classical paradigm). Primary structure refers to a protein's amino acid sequence, which might reveal a cluster of charged residues, say, or a pattern of alternating polar and nonpolar residues. One cannot predict off-hand the conformation of a protein merely from its linear sequence, however, due to rotational flexibility of bonds and non-covalent interactions between non-adjacent amino acids (as well as covalent disulfide bonds).
Physical methods used to interrogate 3D protein structure include X-ray diffraction (XRD), electron microscopy, and nuclear magnetic resonance (NMR) spectroscopy. The paper by Zhang, et al. (1995) that you will refer to today describes the decoding of calmodulin's structure using NMR, which depends on subjecting molecules to electromagnetic fields and analyzing the resulting energy absorption spectra of their nuclei. Scientists who elucidate protein structures, in addition to publishing their results, will often add them to public databases such as the Protein Data Bank (PDB). Because many proteins have structural motifs in common (e.g., alpha helices and beta sheets at the secondary level, or leucine-rich repeats at the tertiary level), which ultimately arise from their amino acid sequences, such databases can be useful for making predictions about proteins with known amino acid sequences but unknown structures. Today we will use a computer program that harnesses information in the Protein Data Bank to display interactive 3D models.
After examining both two- and three-dimensional protein information, you will propose a mutation to the wild-type inverse pericam protein, and finally design primers for incorporating this modification at the genetic level.
References
Chin, D., and A. R. Means. "Calmodulin: A Prototypical Calcium Sensor." Trends Cell Biol 10, no. 8 (August 2000): 322-328.
Zhang, M., et al. "Calcium-Induced Conformational Transition Revealed by the Solution Structure of Apo Calmodulin." Nat Struct Biol 2, no. 9 (September 1995): 758-67.
Protocols
To help you manage your time today, recommended times for the parts below are: Part 1 (30-45 min), Parts 2+3 (45-60 min total), Parts 4+5 (60-90 min total). Some parts may take you shorter or longer, depending on your prior experience with primer design and/or protein modeling.
Part 1: Protein Backbone
Perhaps nothing is so conducive to a feeling of intimate familiarity with a protein as studying it at the amino acid level (primary structure). For the first part of lab today, you will examine a two-dimensional representation of inverse pericam.
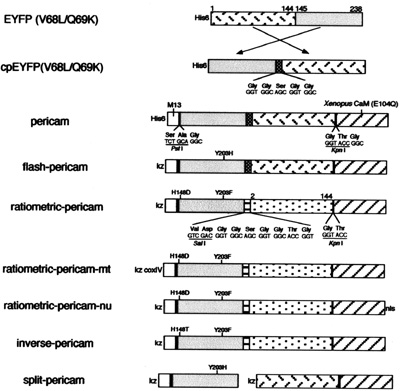
Schematic structures and sequences of pericams for expression in bacteria and mammalian cells. Sequences of linkers and amino acid substitutions are shown below and above the bars, respectively. His-6, the polyhistidine tag; kz, Kozak consensus sequence; nls, nuclear localization signal; coxIV, cytochrome c oxidase subunit IV targeting signal. (Courtesy of National Academy of Sciences, U. S. A. Used with permission. Source: Nagai, T., et al. "Circularly permuted green fluorescent proteins engineered to sense Ca2+." PNAS 98, no. 6 (March 6, 2001): 3197-3202. Copyright (c) 2001 National Academy of Sciences, U.S.A.)
- Begin by downloading this document (PDF), which contains the amino acid and DNA sequences of inverse pericam (IPC). You can compare the annotated sequence file to Figure 1 of the paper by Nagai, et al., which depicts the inverse pericam construct in schematic form. In the sequence file, the M13 peptide is highlighted in magenta, the EYFP sequence in yellow, and calmodulin (CaM) in green. Linker sequences connecting these three parts are shown in blue lettering.
- To help you locate the binding sites for calcium (in the calmodulin portion of IPC), read the following portions of the Zhang paper, along with skimming whatever you find useful: abstract, first two paragraphs, "Linker and loop flexibility" section.
- In your IPC sequence document, mark the amino acid residues that make up the calcium-binding loops in CaM in boldface. To keep yourself oriented, use the fact that the CaM within inverse pericam is an E104Q mutant, that is, the 104th residue of calmodulin is Q.
- Do the four calcium binding loops share any common features? You might imagine that negating or enhancing such features could decrease or increase calcium affinity, respectively.
- If you find other areas of calmodulin that you may be interested in mutagenizing (e.g., hydrophobic pockets), mark these as well. You may find the "Loss of hydrophobic cavities" section in Zhang, et al. helpful.
- Keep in mind that when this module was debuted two years ago, everyone mutated residues directly in the calcium binding loops, and very few groups saw dramatic changes in affinity or cooperativity of calmodulin with respect to calcium. The most interesting mutation results were for D20P, D20R, and G98P. Last year's class-wide results suggested that mutations in the first two binding loops were more likely to have an effect than mutations in the latter two binding loops. Some folks also targeted non-binding structural areas such as T79, but results were inconclusive. You may repeat or otherwise build upon prior results as long as you give your own reasoning.
- Another interesting site to mutate may be the 104 residue itself, as the E104Q mutation was introduced in the first place to alter the calcium response curve of calmodulin from a biphasic to a monophasic one.
- In addition to the E104Q mutation that you already know about, the inverse pericam construct contains a D64Y mutation, which surprisingly is not mentioned by the authors! You might consider targeting this residue or even reversing the mutation.
- Finally, if you're feeling ambitious, you might look into what residues on calmodulin interact with M13, and modify one of them instead. (For example, see the reference by Green, et al. from Prof. Jasanoff's lab.) One group attempted this approach last year and their data was suggestive of a change in calcium-sensing behaviour.
Print out your annotated document and hang on to it for reference. Now let's put some visuals to all those letters!
Reference
Green, D. F., et al. "Rational Design of New Binding Specificity by Simultaneous Mutagenesis of Calmodulin and a Target Peptide." Biochemistry 45, no. 41 (October 17, 2006): 12547-12559. [Full text in PubMed Central]
Part 2: Higher-order Protein Features
Unless we are precocious bioengineers indeed, looking at the amino acid sequence alone is unlikely to tell us too much about the protein. We might be left wondering where the binding sites for M13 and for calcium ions are located in calmodulin, for example. In the previous section, you read some primary scientific literature to locate these features. Now you will use a tool called Protein Explorer to visualize them. As you work, you can ask yourself why these stretches of the protein might work the way that they do, and how they might be changed.
- Protein Explorer is a free web-based viewer for biological molecules. To access it, open the site in your browser. Choose "FirstGlance in Jmol" to proceed.
- Structures are organized according to PDB (Protein Data Bank) identification codes, which may be input at the prompt at the top of the page. Begin by looking at the molecule with PDB ID number 1CLL, which is a calcium-bound form of calmodulin. Later you will search for an example of the ligand-free form, also called apo calmodulin.
- The program will open in FirstView mode for the structure you've chosen (ensure that popup blockers are off if the structure fails to load). On the right is the image panel, which shows your protein along with associated ligands (in this case, calcium). Try clicking and dragging on the rotating image to see what happens.
- Now look at the control panel on the upper left: here you can modify the image. Try adding and removing water molecules and ligands see where they interact with the protein.
- As you explore the features of the control panel and image panel, be sure to observe the message frame window on the lower left for any relevant information that may pop up. If you click on an atom in the image panel, its atomic identity will be displayed in the message frame, along with its encompassing amino acid residue and position.
- From the control panel, click on the PDB icon, which leads to detailed information about the publication upon which the model image is based.
- To find further options for modifying how you view the image, or search for particular atoms, click on More Views in the control panel, or on Jmol at the bottom right of the image panel. For example, you can highlight specific amino acids, or change from a backbone trace to a space-filling model. Explore these features. For example, you might use colour to highlight all the acidic amino acids in calmodulin.
- Be sure to note any useful information in your notebook as you go. You might ask:
- what method was used to elucidate the structure of this protein?
- how good is the image resolution?
- which species did this protein come from?
- when did the authors publish their results?
- what are the major components of the molecule's secondary structure?
- what do the calcium binding loops (or other areas of interest you found) look like?
- Once you are satisfied with your understanding of calcium-bound calmodulin, bring up an apo calmodulin structure (or two) for comparison. You might find the structure directly by using PDB, or by using the NCBI Structure database. Write a few sentences in your lab notebook describing the differences between the calcium-bound and apo forms of calmodulin.
- If you're interested in altering the affinity of the CaM-M13 interaction, you can look at structure 2BBM (or 1CDL to see the interaction of CaM with the full protein from which M13 is derived).
Part 3: Choice of Mutation Site
You will now integrate the information you learned about inverse pericam (especially calmodulin) at the structural and residue levels. Decide on a modification that might plausibly increase or decrease CaM's affinity for calcium (or M13), or might affect cooperativity among the four binding sites. Briefly state your hypothesis for the effect of this mutation in your notebook. Write your mutation as X#Z, where X is the original amino acid, Z is the modified amino acid, and # is the residue number with respect to calmodulin (not IPC as a whole). For example, residue 379 of inverse pericam is residue 101 of calmodulin, so a mutation at that site would be written X101Z.
Note that throughout this module, you will perform most experiments not just with your chosen mutant, but also with a mutant chosen by the teaching faculty that has a known effect, namely M124S. The design examples below use S101L instead because it is a clearer teaching example. You will be given the primer design information for M124S later in the module.
Part 4: Primer Design for Mutagenesis
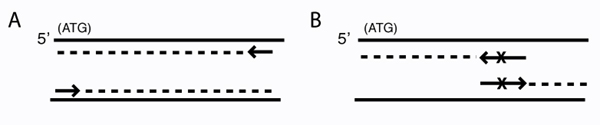
Schematic of primer design for traditional PCR and for mutagenesis. PCR amplification (A) and site-directed mutagenesis (B) are depicted. Arrows represent primers. Solid lines are template DNA, with the coding strand on top. Dashed lines indicate copies of the template. (A) Primers for PCR amplification are perfectly complementary to the template. (Note: the complexity of the first few rounds of PCR is not shown.) (B) Mutagenic primers differ from the template at X. (Note: for circular DNA, the entire template will ultimately be copied.)
It wouldn't be very experimentally efficient for us to somehow pick out and modify a single residue on inverse pericam post-translationally. Instead, we would like to genetically encode our desired mutation, by making mutated copies of a plasmid originally containing inverse pericam DNA. In order to copy DNA, DNA polymerases require short initating pieces of DNA or RNA called primers. Synthetic primers can be used for incorporating desired mutations into DNA, as well as for other purposes, such as non-mutagenic amplification of a specific piece of DNA. For amplification, forward and reverse primers that target the non-coding and coding strands of DNA, respectively, are separated by a distance equal to the length of the DNA to be copied (see figure, part A). In contrast, primer design for site-directed mutagenesis is quite straightforward: both primers are directed at the same location on each strand, and thus will be precisely complementary (see figure, part B). Both direct and mutagenic amplification require cycles of DNA melting, annealing, and extension, which we will discuss more next time.
Good primers must meet several design criteria in order to promote specificity and efficiency of the desired amplification. Length is one important design feature. Primers that are too short may lack requisite specificity for the desired sequence, and thus amplify an unrelated sequence. The longer a primer is, the more favourable are its energetics for annealing (i.e., "sticking") to the template DNA, due to increased hydrogen-bonding pairs. On the other hand, longer primers are more likely to form secondary structures such as hairpins, leading to inefficient template priming. Two other important features are G/C content and placement. Having a G or C base at the end of each primer increases priming efficiency, due to the greater energy of a GC pair compared to an AT pair. The latter decrease the stability of the primer-template complex.
Overall G/C content should ideally be 50 +/- 10%, because long stretches of G/C or A/T bases are both difficult to copy. The G/C content also affects the melting temperature, which should be high for mutagenesis.
In summary, you should heed the following design guidelines:
- The desired mutation (1-3 bp) must be present on both strands.
- The mutation should occur approximately in the middle of the sequence.
- The primer should be 25-45 bp long.
- A G/C content of > 40% is desired.
- Both primers should terminate in at least one G or C base.
- The melting temperature should exceed 78 °C, according to:
- Tm = 81.5 + 0.41 (%GC) – 675/N - %mismatch
- N is primer length, and the two percentages should be whole numbers
As you work, create a step-by-step record of your primer design in a Word document. The logic for the major steps of your selection process should be clear. For example, once you have chosen your mutation site, you might begin by writing out the codon of interest. Next consider the options for coding a new amino acid, using this Genetic Code table from New England Biolabs (NEB). Finally, you might pick the codon that incurs the fewest point mutations. An example is outlined below.
Residue 101 of calmodulin is serine, encoded by AGC. This is residue 379 with respect to the entire inverse pericam construct, and we can find it and some flanking code in the DNA sequence from Part 1:
|
361 (5') GAG GAA ATC CGA GAA GCA TTC CGT GTT TTT GAC AAG GAT GGG AAC 376 (5') GGC TAC ATC AGC GCT GCT CAG TTA CGT CAC GTC ATG ACA AAC CTC To change from serine to leucine, one might choose UUA, UUG, or CUX (wherer X=U, A, G, or C). Because CUC requires only two mutations (rather than three as for the other options), we choose this codon. Now we must keep 15-20 bp of sequence on each side in a way that meets all our requirements. To quickly find G/C content and see secondary structures, look at IDT's OligoAnalyzer Web site. (Note that the Tm listed at this site is not one that is relevant for mutagenesis.) Ultimately, your primer and its complement might look like the following, which has a Tm of almost 81, and a G/C content of ~58%. 5' GG AAC GGC TAC ATC CTC GCT GCT CAG TTA CGT CAC G 3' C GTG ACG TAA CTG AGC AGC GAG GAT GTA GCC GTT CC |
Next you will modify your primers further according to Part 5, if possible for your mutation.
Part 5: Additional Silent Mutation
One goal of this module is to interrogate DNA sequences in multiple ways. To set us up for achieving this goal, you should attempt to incorporate a second mutation in inverse pericam using your primers. This time, the purpose of the mutation is not to change the protein, but to create a recognizable DNA tag. Thus, the mutation must be silent, that is, it should not affect the protein code. For example, CCA to CCG is a silent mutation, because both triplets code for the amino acid Proline. You can use the NEB Genetic Code table to find degenerate codons.
One category of useful DNA tags are called restriction sites. These sequences, usually short and palindromic, are recognized by enzymes that cut the sites in unique and specific ways, as we will discuss further next time. For now, all you need to know is that you would like to create a new restriction site in your DNA, ideally one that exists in only one or two (or zero) other places in the entire inverse pericam plasmid. All these requirements are getting pretty complicated, and you can imagine that they become quite time-consuming to satisfy by hand! We will use some freely available computer programs to help us.
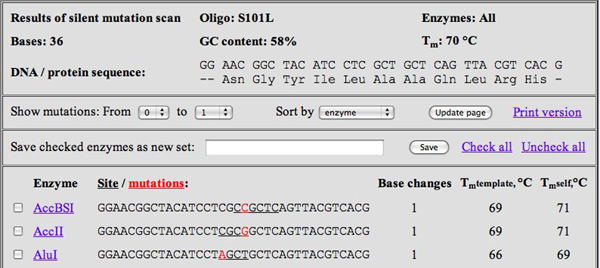
WatCut Analysis screenshot for S101L primer. (Courtesy of Michael Palmer. Used with permission.)
- At the WatCut Web site, created by Michael Palmer at Waterloo University, click on Silent Mutation Analysis and input the primer you designed in part 4. After choosing the appropriate reading frame (i.e., make sure the amino acids are correct!), what you receive back will be a list of restriction sites that result from making specific point mutations, which themselves are highlighted in red lettering. For example, for the S101L primer shown above, about 55 choices come up (see screenshot figure).
- Start out by looking at the results for 0 to 1 mutations. Some of you may have the lucky result that your original (non-silent) mutation creates a new restriction site, and you would miss this if you did not look at the 0 mutation results.
- Print out the results page for your mutation (and for S101L (PDF) if you want to practice (S101L file courtesy of Michael Palmer, used with permission). You should also use the New England Biolabs NEBcutter tool to produce a list of restriction sites that occur 1-2 times in the inverse pericam plasmid, and those that cut 0 times.
- On your results page, cross off any choices that do not occur on either of the restriction site lists. For any choice that is on one of the lists, write a "2" or "1" or "0" next to it, depending on how many times it is present in the plasmid. This should leave very few choices indeed! For example, for the S101L primer, only two choices is left: BsgI and FspI, both double-cutters.
- If you have more than one choice left, you can take other considerations into account. For example, ideally the silent mutation should be close to the middle of the primer, just like your non-silent one, but mutations farther away often work as well. This criterion does not distinguish between our S101L choices.
- For a double-cutter, another consideration is how far apart the two sites are. For BsgI, they are at ~400 and 700 base-pairs, or only 300 bp apart. For FspI, the sites are at ~1700 and 2900, or 1200 bp apart. A larger spacing is generally advantageous for later analysis.
- If you are still unsure of which mutation to choose, or if you are left with zero choices, ask the teaching faculty for assistance.
Keep in mind that some stretches of DNA simply may not lend themselves to incorporating a new restriction site via silent mutation. You will still have the opportunity to analyze the M124S candidate for all subsequent experiments, and you can do most experiments with your single-mutation candidate as well.
Along with your notebook today, you should hand in your primer design record, and annotated printout from the Watcut Web site. The final primer should be annotated as below:
|
GG AAC GGC TAC ATC CTC GCT GCG CAG TTA CGT CAC G The underlined codon was changed from AGC (Ser) to CTC (Leu). It is residue 379 of inverse pericam and residue 101 of calmodulin. The point mutation in bold creates the new restriction site FspI, TGCGCA, which begins within residue 381 of inverse pericam. |
Finally, input your primers in today's results table. This primer and its reverse complement will be ordered for you in time for the next class.
For Next Time
- Look ahead to Part 3 (Journal Article discussion) of our next lab class, and begin reading the two papers we will discuss in class next time. Because you had a lot of work due today, you will also have some in-class time on Day 2 (~45 min, say) to finish your close reading of the article. There is no need to hand in written answers to any of the questions - they are simply meant to guide your reading.