
Modules: 1.1 | 1.2 | 1.3 | 1.4 | 1.5 | 1.6 | 1.7
Introduction
Today you will ligate your linearized M13KO7 backbone with your oligonucleotide insert by mixing the two in the presence of ATP and an enzyme, T4 DNA ligase. During the ligation reactions, hydrogen bonds will form between the overhangs on the fragments, and then the ligase will repair the phosphate backbone, creating a stable circular plasmid
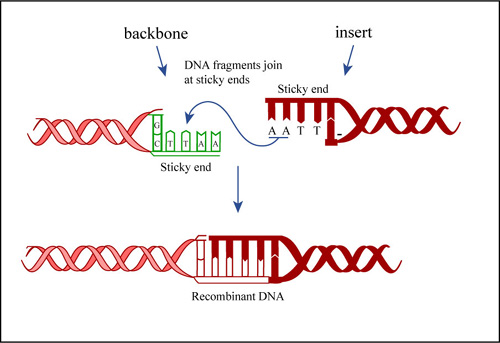
Ligation Reactions. (Figure by MIT OpenCourseWare.)
DNA Ligation
Hopefully, your ligation reactions will generate your desired construct, namely the M13KO7 backbone carrying a short added sequence in the gene for p3. Alternative ligation products may arise, including a simple reclosing of the singly cut M13KO7 backbone. We will remove these easy to form but undesired products with a trick called a "kill cut," described below.
You will transform today's ligation reactions into bacteria. During 'transformation,' a single plasmid from the ligation mixture enters a single bacterium and, once inside, replicates and expresses the genes it encodes. One of the genes on the M13KO7 genome leads to kanamycin-resistance. Thus, a transformed bacterium will grow on agar medium containing kanamycin. Untransformed cells will die before they can form a colony on the agar surface.
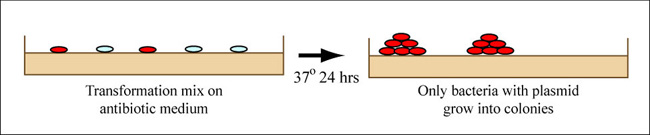
Growing colonies on medium, showing that only bacteria with plasmid grow into colonies. (Figure by MIT OpenCourseWare.)
Bacterial Transformation
Most bacteria do not usually exist in a 'transformation ready' state, but the bacteria can be made permeable to the plasmid DNA, and cells that are capable of transformation are referred to as 'competent.' Competent cells are extremely fragile and should be handled gently, specifically kept cold and not vortexed. The transformation procedure is efficient enough for most lab purposes, with efficiencies as high as 109 transformed cells per microgram of DNA, but it is important to realize that even with high efficiency cells only 1 DNA molecule in about 10,000 is successfully transformed.
Protocols
When you have a break from the work described below, be sure to examine the plaques you plated last time. Record the number of plaques on each plate, their appearance and any observations or conclusions you can draw.
Part 1: Anneal Oligonucleotide Insert
First, you will anneal the insert you designed last time, making a small double stranded fragment of DNA that has sticky ends compatible to the ones in the M13KO7 backbone.
- Check the oligonucleotides that were ordered for you by examining the information sheets sent to you by the company. Are the oligos the correct sequence? If they are right, then you should resuspend the samples in sterile water, using the listed number of nmoles to calculate how much sterile water you should add for a final concentration of 100 pmoles/µl. Give the tubes a quick spin in the microfuge to bring down any material that is stuck on the lid. Add sterile water to resuspend the oligos at a concentration of 100 pmoles/µl, and write this concentration on the spec. sheets from the company, then turn the sheets back to the teaching faculty.
- You will anneal the oligos in a PCR tube that you can get from the faculty. These are very small (relative to eppendorf tubes) and fit nicely into the holes of the yellow pipet tips boxes. Label the top or side of one tube as best you can with your team ID and the contents of the tube.
- Mix 10 µl of each of the appropriate oligonucleotides in the PCR tube.
- Add 11 µl of 10X T4 DNA ligase buffer from New England Biolabs. This buffer will provide the salts necessary to stablize the annealed primers, and will be consistent with the conditions needed for ligation (next time).
- Heat the sample to 94° for 2 minutes in the lab's thermal cycler ("PCR machine") and then let it cool slowly.
- The teaching faculty will store any remaining oligo solutions that you have not annealed.
Part 2: Ligation reactions
For your ligation, you will mix the M13KO7 backbone you prepared with the annealed oligonucleotides. As control reactions you will also prepare a 'bkb, no ligase' reaction to control for any errant uncut plasmid that might have wandered into your solutions. Additionally you will prepare 'bkb only, plus ligase' reaction to assess the frequency of backbone religation despite the "kill cut."
The contents of each ligation will be
| bkb, no ligase | bkb only, plus ligase | bkb + insert, plus ligase | |
|---|---|---|---|
| M13KO7 bkb | 4 μl | 4 μl | 4 μl |
| Insert | None | None | 5 μl |
| 10X Ligation Buffer^ | 1.0 μl | 1.0 μl | 1.0 μl |
| T4 DNA Ligase | None | 0.5 μl | 0.5 μl |
| Water | To 10 µl not Including Volume of Enzyme | ||
^New England Biolabs sells 10X Ligation buffer to use with their ligase. It contains ATP so must be kept on ice.
- Assemble the reactions in eppendorf tubes but not in the order listed. Please ask if you are unsure what order to assemble the components or what must be kept on ice.
- When the ligation mixes are complete, flick the tubes to mix the contents, quick spin them in the microfuge to bring down any droplets, then incubate the reactions at room temperature for at least 10 minutes.
- Recall that the ligation of your insert into the M13KO7 backbone will destroy the BamHI restriction sites originally used to linearize the genome. Consequently, you can enrich for the proper ligation products by digesting the ligation reactions with the enzyme used to open up the backbone. These are called "kill cuts" since in theory they destroy unwanted ligation products. So while your samples are ligating, you should prepare a "killcut" cocktail according to the table below. By making one mixture that contains water, buffer and enzyme (commonly called a reaction "cocktail"), you can add the same mixture to all reactions, minimizing effects of pipet-error and possible accidental errors, like leaving one component out of one reaction that you remember in all the others. You will prepare enough cocktail to perform 4 killcut reactions, even though you have only three. This will assure you have enough volume for the three reactions.
Volume information.
volume in each reaction x4 = volume in cocktail Water 8 µl ? µl 10X NEB buffer 2 µl ? µl Enzyme 0.25 µl BamHI ? µl
Add 10 µl of the killcut cocktail to each of the ligation reactions you've prepared, pipetting up and down to mix. - Incubate at 37° for 15 minutes.
Part 3: Precipitation of DNA
In this step, salts and buffers are removed from the reactions. DNA is precipitated with salt and ethanol. Yeast tRNA is added to the precipitation as "carrier," allowing you to better visualize the DNA pellets. The salts are washed from the pellets with 70% ethanol. The tRNA is not removed. Rather it enters the bacteria with the ligation DNA, but is then rapidly degraded.
- Add 20 µl 3M sodium acetate to each tube.
- Add 5 µl tRNA to each tube.
- Add 200 µl cold 100% ethanol to each tube and vortex.
- Spin in a room temperature microfuge 15 minutes. Be sure to orient your tubes in the microfuge so you know where your pellets should be and balance your tubes with those of another group or with a water-filled eppendorf tube.
- When the spin is done, locate the pellets in each of the eppendorf tubes. They may appear as solid white dots at the bottom corner of the tube or they may appear to be a diffuse white smear along the wall of the tube. Both are OK. Carefully remove the ethanol from the pellets with your P1000, taking care not to disturb the pellet. You do not have to remove every last drop.
- Wash the pellets with 500 μl cold 70% ethanol. This is done by dribbling the 70% ethanol along the wall of the eppendorf tube that is opposite your pellet and then removing the 70% ethanol with the same pipet tip. Again you should not disturb the pellet and you do not have to remove every drop of liquid in the tube. If the pellet seems to float away from the wall of the tube, you can re-spin the tubes for 2 minutes with the liquid to adhere the pellets to the wall again.
- Once you have washed all your pellets, give the tubes a quick spin in the microfuge to bring down any droplets of ethanol that cling to the sides of the tube then remove any remaining liquid from the tubes using your P200. Allow your tubes to dry in the hood for 10 minutes. All the ethanol must be removed or evaporated.
- Resuspend the pellets in 15 μl sterile water. This is done by adding water to the tubes and mixing. If the DNA does not readily go into solution, it helps to heat the DNA in the 42°C heat block, then vortex and pipet up and down several times. Bring any droplets down to the bottom of the tubes with a quick spin in the microfuge.
Part 4: Bacterial transformation
You will perform 4 bacterial transformations, one for each of the three ligation mixtures as well as one transformation with 5 ng of plasmid DNA to assess transformation frequency.
- Prewarm and dry five LB+Kan plates by placing them in the 37°C incubator, media side up with the lids ajar. You really will need five plates even though you are only doing four transformations, since your "backbone + insert" sample will be plated twice.
- Get an aliquot of competent cells from one of the teaching faculty. Keep these cells on ice at all times. There should be at least 200 μl of cells in each tube. Aliquot 50 μl of cells into 4 clean eppendorf tubes.
- Add DNA to each tube of cells as shown in the table below.
- Flick to mix the contents and leave the tubes on ice for at least 5 minutes.
- Heat shock the cells at 42°C for 90 seconds exactly. Use your timer.
- Move the samples to a rack on your bench then use your P1000 to add 0.5 ml of LB media to each eppendorf tube. Invert each tube to mix.
- Incubate the tubes in the 37°C incubator for at least 30 minutes. This gives the kanamycin-resistance gene some time be expressed in the transformed bacterial cells.
- While you are waiting, label 4 large glass test tubes with your team color and numbers 1, 2, 3, 4. Mix 10 ml LB with 10 µl of the Kanamycin stock. Aliquot 2.5 ml/tube. This will help the teaching faculty to set up overnight cultures for you for next time.
- Plate 200 µ of each transformation mix on LB+Kan plates, plating the bkb+insert+ligase transformation twice. Note: After dipping the glass spreader in the ethanol jar, then pass it through the flame of the alcohol burner just long enough to ignite the ethanol. After letting the ethanol burn off, the spreader may still be very hot, and it is advisable to tap it gently on a portion of the agar plate without cells in order to equilibrate it with the agar (if it sizzles, it's way too hot). Once the plates are done, wrap them with colored tape and incubate them in the 37°C incubator overnight. One of the teaching faculty will remove them from the incubator and set up liquid cultures for you to use next time.
Volume information.
Tube Transformation Add 1 Positive control plasmid 1 µl (5 ng) of M13KO7 DNA 2 bkb, no ligase 5 µl 3 bkb, plus ligase 5 µl 4 bkb+insert, plus ligase 5 µl
DONE!
For Next Time
Questions 1 and 2 are theoretical but they should help prepare you to interpret the results you will collect next time
- You have purchased some supercompetent bacteria that are provided at a transformation efficiency of 109 colony forming units/ug of DNA. You transform the cells with 1 ng of plasmid DNA and plate 1/1000th of the cells. How many colonies do you expect? Next you transform another aliquot of cells, also at 109 colony forming units/ug of DNA, with 2 µl of plasmid DNA. You spread 1/100th of the cells and find 50 colonies growing on the plate after 24 hours at 37°C. What is the concentration of plasmid?
- To illustrate your understanding and the importance of the controls you performed today, please write a one-sentence interpretation for each of the following transformation outcomes.
- Outcome 1: no colonies on any plate.
- Outcome 2: thousands of colonies on all the plates.
- Outcome 3: approximately the same number of colonies on the backbone+ligase+kill cut as the backbone+insert+ligase+kill cut.
- There may be more than one valid interpretation for some of the data (only one answer for each is required for the assignment).
- Next time you will isolate DNA from four transformants and begin to characterize the plasmids in these bacteria. To prepare for this experiment, you should draw a plasmid map of the M13KO7 genome. Start by printing out the M13KO7 plasmid map from NEB by using their NEB Cutter tool, selecting M13KO7 from the "Viral and phage" drop down menu on the right, changing the default minimum ORF to 25 amino acids (do you remember which of the M13 proteins are very small?), and finally telling the program that you are entering circular DNA. Modify the map by hand to indicate which restriction site you are changing, which enzymes you are adding, and how many basepairs of DNA this modification needs. Next, use the plasmid map to help you plan at least two restriction digests that will confirm the presence of the oligonucleotide insert. Recall that the lab does not have every enzyme available so you should double check your idea against the list of available enzymes. It will help to read the introduction for the next lab before you complete this part of the assignment. Be sure to predict the size of the fragments you expect when the plasmid does and doesn't have the oligonucleotide insert. Also include reaction conditions such as buffer and temperature. Use the NEB Web site for details on various enzymes and reaction conditions.
Lecture notes files.
Plasmid with insert Plasmid no insert Diagnostic digest 1 Enzyme(s) used Buffer used Temperature Predicted fragments Diagnostic digest 2 Enzyme(s) used Buffer used Temperature Predicted fragments - Based on the results of your plaque assay, what is the titer of each stock solution of phage? Please show your work. If the plaques appeared different, please consider how the phage genomes differ (M13KO7 is a "helper phage" while E4 is identical to the M13 genome except four glutamic acids are presented on the N-terminus of the p8 protein) and suggest how these differences might account for the differences in plaque morphology.
- Read the article by Chan, Kosuri and Endy. "Refactoring bacteriophage T7" Nature/EMBO Molecular Systems Biology 13 September 2005 doi:10.1038/msb4100025 and News & Views. Come prepared to discuss this paper during lab next time. To guide your reading and test your understanding, try to answer the following questions (note: these questions are just to guide your reading and the answers do not have to be turned in):
- from the Introduction:
- What is "refactoring" and what makes is T7 an attractive candidate for this approach?
- What experimental techniques give us "component level" understanding, i.e. allow us to attribute particular functions to particular sequences in the genome? How completely can "component level" understanding provide "system level" understanding?
- How predictive have computational and quantitative models for T7 behavior proven to be? What's important about predicting behavior?
- from the Results:
- What design principles were the authors pursuing? How well do these map to our class effort at M13 re-design?
- Was the entire T7 genome refactored?
- What techniques were used to verify the refactoring? What techniques were used to evaluate it?
- from the Discussion:
- How do the authors' findings extend knowledge of T7 biology?
- Does T7.1 resolve disagreements between model-based behavior predictions and those that are observed though experimental approaches?
- Could nature have produced the T7 phage that now exists in the Endy lab in Building 68?
- What's next for this phage?
- from the Introduction:
Reagents list
- 5 ng M13KO7 DNA
- T4 DNA Ligase Buffer (1X)
- 50 mM Tris-HCl
- 10 mM MgCl2
- 10 mM DTT
- 1 mM ATP
- 25 μg/ml BSA
- LB
- 1% Tryptone
- 0.5% Yeast Extract
- 1% NaCl
- LB+kan plates
- LB with 2% agar and 25 µg/ml Kanamycin